Neurologie
Weitere neurologische Beschwerden und Erkrankungen
Creutzfeldt-Jakob-Krankheit
Creutzfeldt-Jakob-Krankheit (CJK): Sehr seltene, aber immer tödlich verlaufende Gehirnerkrankung, die durch einen raschen Verfall der geistigen und körperlichen Fähigkeiten gekennzeichnet ist. Die Erkrankung gehört zu den spongiformen Enzephalopathien, bei denen das Gehirngewebe unter dem Mikroskop typische "Löcher" zeigt (spongiform = schwammartig). Ursache sind fehlgefaltete Proteine (pathologische Prionproteine), die im Gehirn zum Untergang der Nervenzellen führen. Die Erkrankung tritt in den meisten Fällen sporadisch auf, nur bei jedem Zehnten ist sie genetisch verursacht. CJK kann auch durch Transplantation von Geweben Erkrankter übertragen werden. Bei einer seit 1995 bekannten Variante der CJK (nVCJK) infiziert sich der Mensch mit den pathologischen Prionproteinen, indem er Fleisch von an Rinderwahn (BSE) erkrankten Rindern isst.
Alle Formen der CJK sind unheilbar. Ist die Erkrankung einmal ausgebrochen, versterben die Betroffenen meist innerhalb von Monaten.
Symptome und Leitbeschwerden
- Rasch fortschreitender Abbau der geistigen Fähigkeiten
- Persönlichkeitsveränderungen, z. B. Reizbarkeit, Verstimmung, Ängstlichkeit, Wahnvorstellungen
- Schlaflosigkeit
- Verlust von geistigen Fähigkeiten, beginnend mit Gedächtnisstörungen und fortschreitend bis zur Demenz
- Sehstörungen
- Bewegungsstörungen, z. B. Koordinationsstörungen (Bewegungen gehen am Ziel vorbei), Muskelzuckungen, Gangunsicherheit
- Bewegungslosigkeit und Sprachlosigkeit in der Endphase (akinetischer Mutismus).
Wann zum Arzt
In den nächsten Tagen, wenn
- sich oben genannte Beschwerden entwickeln.
Die Erkrankung
Verursacht wird die Creutzfeldt-Jakob-Krankheit durch Prionen, also durch krankmachende Eiweiße, welche sich von gesunden Eiweißen im Gehirn nur durch ihre räumliche Anordnung unterscheiden und die die gesunden Eiweiße auf noch nicht geklärte Weise in die krankhafte Form umwandeln. Durch die Anreicherung der abnormen Eiweiße sterben die Nervenzellen im Gehirn nach und nach ab, wobei die Beschwerden unaufhaltsam fortschreiten und nach wenigen Wochen bis Jahren zum Tod des Patienten führen.
Formen
Bei der CJK werden je nach ihrer Ursache verschiedene Formen unterschieden.
Sporadische CJK oder sporadische Prionerkrankung (etwa 90 % der CJK-Fälle). Diese Form ist mit etwa 1–1,5 Fällen pro 1 Million Einwohner die häufigste Form der CJK, in Deutschland werden jährlich etwa 100 Fälle gemeldet. Die Patienten erkranken zwischen dem 60. und 70. Lebensjahr, auslösende Faktoren sind nicht bekannt. Typisch ist die schnelle Verschlechterung mit rasch fortschreitender Demenz, Bewegungsstörungen (Ataxie) und Muskelzuckungen. Am Ende kommt es zum sog. akinetischen Mutismus, der Patient bewegt sich nicht mehr, spricht nicht mehr und nimmt an nichts mehr teil. Die Betroffenen versterben meist innerhalb von 4 bis 6 Monaten.
Familiäre CJK oder genetische Prionerkrankung (etwa 10 % der CJK-Fälle). Diese familiäre CJK wird autosomal-dominant vererbt, sie tritt um das 50. Lebensjahr herum auf, d. h. etwas früher als die sporadische CJK. Es sind mehrere molekulare Mutationen bekannt. Typisch ist die zunehmende Gangunsicherheit bei den Patienten, die Demenz bildet sich erst im späteren Verlauf der Erkrankung aus.
Übertragene, iatrogene CJK. Diese Sonderform wird durch ärztliche Prozesse (d. h. iatrogen) von Mensch zu Mensch übertragen, und zwar über direkten Kontakt mit infektiösem Gewebe oder kontaminierten neurochirurgischen Instrumenten. So kam es beispielsweise nach Transplantation von Augenhornhaut oder Hirnhaut verstorbener, unerkannt an CJK erkrankter Spender bei den Organempfängern zu CJK. Bevor Wachstumshormon (STH) gentechnisch hergestellt wurde, führte auch das bei Wachstumshormonmangel früher übliche Spritzen von STH aus Leichenhypophysen zu einigen CJK-Fällen.
Übertragene, neue Variante der CJK (nVCJK). Seit 1995 wurden in Großbritannien und anderen Ländern über 200 Fälle einer neuen Variante der Creutzfeldt-Jakob-Krankheit (nvCJK) beobachtet. Die Erkrankten sind deutlich jünger, die Beschwerden etwas anders gewichtet. So stehen zu Beginn der Erkrankung psychiatrische Auffälligkeiten wie Depression oder Psychose im Vordergrund, später kommen Missempfindungen und Gangunsicherheiten und am Ende die Demenz dazu. Die Krankheit verläuft insgesamt etwas langsamer als die sporadische CJK, die Betroffenen versterben nach etwa 14 Monaten.
Nach heutigem Kenntnisstand ist diese neue Variante der Creutzfeldt-Jakob-Krankheit die menschliche Form von BSE, der bovinen spongiformen Enzephalopathie, besser bekannt als Rinderwahnsinn. Übertragen wird die Erkrankung höchstwahrscheinlich vor allem durch den Verzehr des Fleischs erkrankter Rinder, denn durch Kochen werden die Prionen nicht abgetötet. Auch Infektionen durch Blutübertragung sind bekannt geworden
Nach neuen Vorschriften und Auflagen für die Verarbeitung von Rindern sind BSE-Fälle in der EU drastisch zurückgegangen.
Diagnosesicherung
Eine sichere Diagnose ist nur durch Untersuchung des Gehirngewebes nach dem Tode möglich. Als Hinweise auf die Creutzfeldt-Jakob-Krankheit zu Lebzeiten gelten eine schnell fortschreitende Demenz zusammen mit den oben genannten Symptomen. Zusätzlich wird eine Elektroenzephalografie (EEG) durchgeführt, und die Gehirnflüssigkeit (Liquor) auf spezielle Proteine überprüft. Auch eine Kernspin-Untersuchung trägt zur Festigung der Verdachtsdiagnose bei, mit ihrer Hilfe lassen sich die schwammartigen Veränderungen in der Hirnsubstanz nachweisen.
Differenzialdiagnosen. Demenz und Bewegungsstörungen kommen in unterschiedlichen Ausmaßen z. B. auch bei Alzheimer-Demenz, Huntington-Krankheit, Hirnentzündungen oder Morbus Wilson vor.
Behandlung
Eine ursächliche Behandlung gibt es nicht, es können nur die Beschwerden gelindert werden.
Myoklonien, also unwillkürliche Muskelzuckungen, sprechen in der frühen Krankheitsphase häufig auf die Gabe von Antiepileptika wie Clonazepam oder Valproat an. Der geistige Abbau kann in Einzelfällen mit der Einnahme von Flupiritin gebremst, aber nicht aufgehalten werden. Das Antibiotikum Doxycyclin soll, wenn es sehr früh eingesetzt wird, die Überlebenszeit verdoppeln, der Wirkmechanismus dafür ist unbekannt.
Prognose
Nach Ausbruch der Erkrankung kommt es rasch zum Tod. Bei der häufigsten Form, der sporadischen CJK, versterben die Patienten meist innerhalb von 4 bis 6 Monaten. Die mit dem Rinderwahnsinn in Zusammenhang gebrachte neue Variante der CJK verläuft etwas langsamer, hier sterben die Patienten durchschnittlich 14 Monate nach Ausbruch der Krankheit.
Weiterführende Informationen
Informationsbroschüre für Angehörige von CJK-Patienten der Göttinger Prionforschungsgruppe (Uniklinik Göttingen) http://cjd-goettingen.de/fileadmin/media/downloads/informationsbroschuere-fuer-angehoerige.pdf
Thema BSE samt Maßnahmenpaket zum Eindämmen der Seuche auf der Webseite des Bundesministeriums für Ernährung und Landwirtschaft: https://www.bmel.de/DE/Tier/Tiergesundheit/Tierseuchen/_texte/BSE.html
Informative Webseite der World Organisation for Animal Health zum Thema BSE (auf Englisch): https://www.oie.int/animal-health-in-the-world/official-disease-status/bse/
Hirndrucksteigerung
Hirndrucksteigerung: Krankhafter Anstieg des Drucks im Schädelinneren mit nachfolgender Schädigung des Gehirns. Die Hirndrucksteigerung kann plötzlich auftreten (z. B. nach schwerer Kopfverletzung) oder sich allmählich entwickeln (etwa infolge eines Gehirntumors). Dementsprechend variieren auch die Beschwerden, bei akuter Zunahme des Drucks stehen Kopfschmerzen, Übelkeit und Bewusstseinsstörungen bis hin zum Koma im Vordergrund. Die chronische Hirndruckerhöhung zeigt sich vor allem durch eine sich allmählich entwickelnde Antriebsstörung. Werden durch den erhöhten Druck lebenswichtige Hirnzentren eingeklemmt, kommt es zu Spastik, lichtstarren Pupillen, Überstreckung des Rumpfes, Schnappatmung und Atemlähmung.
Neben einer evtl. erforderlichen Drucksenkung durch Medikamente oder operative Maßnahmen steht vor allem die Behandlung der Ursache im Vordergrund. Daneben wird die Patent*in intensivmedizinisch überwacht und stabilisiert.
Die Prognose hängt von der Ursache der Hirndrucksteigerung ab. Nach einem unfallbedingten Schädel-Hirn-Trauma ohne weitere Gehirnverletzungen kommt es in vielen Fällen zur Rückbildung der Schwellung und kompletter Ausheilung. Hat sich eine Einklemmung mit Spastik, lichtstarren Pupillen und Atemstörungen entwickelt, sind schwere Dauerfolgen bis hin zum Wachkoma häufig.
Symptome und Leitbeschwerden
- Bei Entstehung innerhalb von Stunden: Unruhe, rasch zunehmende Bewusstseinstrübung, Bewusstlosigkeit
- Bei Entstehung innerhalb von Tagen: Kopfschmerzen, Übelkeit, morgendliches Erbrechen (mit nachfolgender kurzzeitiger Besserung der Beschwerden), Verwirrtheit, zunehmende Bewusstseinstrübung bis zur Bewusstlosigkeit
- Bei Entstehung innerhalb von Wochen und Monaten: Zunächst Verlangsamung, Antriebsstörungen, Verhaltensänderungen, dann Kopfschmerzen, Übelkeit und die oben aufgeführten Beschwerden.
Wann zur Arztpraxis
Sofort die Notärzt*in rufen bei
- Bewusstlosigkeit, rasch zunehmender Bewusstseinseintrübung.
Am gleichen Tag, bei
- Verwirrtheit, Kopfschmerzen, Druckgefühl im Kopf und starker Übelkeit.
In den nächsten Tagen, wenn
- immer wieder Kopfschmerzen und Übelkeit auftreten
- unerklärliche Persönlichkeitsveränderungen auffallen.
Die Erkrankung
Der Schutz des Gehirns durch die Schädelknochen hat seinen Preis: Jede nennenswerte Volumenzunahme, sei es durch eine Blutung, eine entzündungsbedingte Schwellung oder einen Tumor, führt zu einer Drucksteigerung, da eine Volumenausdehnung (wie z. B. bei einer Beule) nicht möglich ist.
Der normale Druck im Schädel liegt im Bereich von 5–15 mmHg. Er ist definiert als der Druck, der vom Schädelinhalt auf die Hirnhaut ausgeübt wird. Bei ansteigendem Hirndruck wird zunächst das geringe Reservevolumen ausgeschöpft, indem die liquorgefüllten Hohlräume zusammengepresst werden. Dann wird das Gehirn zunehmend komprimiert. Dadurch sinkt die Durchblutung, was die Nervenzellen schädigt. In dieser Phase machen sich die oben genannten allgemeinen Hirndruckzeichen wie Kopfschmerzen, Übelkeit, vermehrte Schläfrigkeit bis hin zum Koma bemerkbar.
Steigt der Druck weiter an, werden schließlich Gehirnanteile auch in Richtung Schädelbasis gedrückt und dabei lebenswichtige Zentren eingeklemmt und beeinträchtigt. Diese Einklemmung zeigt sich durch krankhafte Bewegungsmuster, vor allem Streckbewegungen der Arme und Beine, Reflexstörungen und schließlich Beeinträchtigung von Temperaturregulation, Herz-Kreislauf-Funktion bis hin zu Schnappatmung und Atemlähmung.
Ursachen
Folgende Störungen oder Erkrankungen können durch Zunahme des Hirnvolumens oder die Vermehrung von Liquor den Hirndruck erhöhen:
- Schädel-Hirn-Trauma
- Hirnödem aufgrund von Vergiftungen oder Entzündungen (Gehirnentzündung, Hirnhautentzündung)
- Raumfordernde Prozesse wie Gehirntumor, Subduralhämatom
- Hydrozephalus bei Zunahme des Liquors, z. B. durch Abflussstörungen oder eine Überproduktion
- Sinusvenenthrombose.
Diagnosesicherung und Behandlung
Die Betroffenen werden sofort auf die Intensivstation eingeliefert und dort parallel zur Diagnostik engmaschig überwacht. Essenziell ist dabei die Unterstützung von Herz und Kreislauf sowie gegebenenfalls die künstliche Beatmung, um das Gehirn mit möglichst viel Sauerstoff zu versorgen. Außerdem überprüfen und optimieren die Ärzt*innen wichtige Blutwerte wie z. B. die Elektrolyte, den Blutzucker und den Blutdruck.
Die Ursache der Drucksteigerung ermitteln die Ärzt*innen mittels CT und Kernspin. Den Hirndruck selbst kann man nur mit invasiven Methoden messen. Dazu führen sie entweder einen Katheter mit Druckaufnehmer in einen der Hirnventrikel ein. Eine andere Methode ist die Druckmessung direkt im Hirngewebe, bei der eine kleine Sonde mit Drucksensor über ein etwa 15 mm tiefes Bohrloch durch den Schädelknochen eingeführt wird. In manchen Fällen bleibt diese Sonde zur kontinuierlichen Überwachung des Hirndrucks bis zur Erholung der Patent*innen liegen.
Daneben versuchen die Ärzt*innen, den erhöhten Hirndruck zu senken. Angestrebt werden dabei Werte unter 20 mmHg. Um dies zu erreichen, sind je nach Ursache der Druckerhöhung folgende Maßnahmen möglich:
- Therapie der Grunderkrankung, z. B. die Entfernung eines Tumors
- Liquordrainage über eine Punktionskanüle nach außen
- Osmotherapie mit der intravenösen Gabe von hypertoner Kochsalzlösung oder Mannitol. Hierbei soll durch den erhöhten osmotischen Druck im Blut die Flüssigkeit aus dem Gehirn "herausgezogen" und schließlich über die Niere ausgeschieden werden. Aufgrund der Nebenwirkungen (Blutdruckabfall, Entwässerung auch der gesunden Hirnsubstanz, Nierenschädigung) wird diese Therapie nur bei Druckspitzen und unter engmaschiger Kontrolle empfohlen
- Oberkörperhochlagerung um 15–30°
- Eventuell Kortison (z. B. bei Hirntumor oder bakterieller Hirnhautentzündung, nicht aber bei Schädel-Hirn-Trauma)
- Selten Kraniektomie, d. h. Entfernung eines Stücks der Schädeldecke, um den Hirndruck zu senken (umstritten, nur wenn andere drucksenkende Maßnahmen nicht wirken).
Prognose
Der weitere Krankheitsverlauf hängt einerseits von der Ursache der Drucksteigerung ab. Andererseits sind die Aussichten der Patient*innen umso schlechter, je länger der Hirndruck bestand und je höher er war. Überlebt die Patient*in eine Einklemmung, sind schwerste Dauerfolgen bis hin zum Wachkoma die Regel.
Lähmungen
Lähmung: Unfähigkeit, einzelne Körperteile oder ganze Bereiche des Körpers zu bewegen. Typischerweise ist sie Folge einer Schädigung der Muskeln des betroffenen Körperteils oder der sie versorgenden Nervenzellen oder Nerven.
Eine Lähmung ist keine Krankheit, sondern ein Symptom. Mögliche Lähmungsursachen reichen von Gehirnschäden, die ein Kind im Mutterleib erleidet (infantile Zerebralparese) über Schädel-Hirn- oder Rückenmarkverletzungen durch Unfälle bis hin zu Autoimmunerkrankungen und Vergiftungen.
Behandlung und Prognose der Lähmung richten sich nach der zugrundeliegenden Ursache. Bei bleibender Lähmung ist die Rehabilitation von großer Bedeutung. Gegen Folgeschäden wie Muskelschwund und Wundliegen helfen unterstützende Maßnahmen wie Physiotherapie, Massagen oder Elektrobehandlungen.
Symptome und Leitbeschwerden
- Unmöglichkeit, ein Körperteil zu bewegen
- Unfähigkeit, zu sprechen oder zu schlucken.
Wann zum Arzt
Noch am gleichen Tag, wenn
- Lähmungserscheinungen auftreten – auch wenn sie von selbst wieder weggehen – sie können Warnzeichen z. B. eines drohenden Schlaganfalls oder einer Hirnaneurysmablutung sein.
Sofort zum Arzt oder ins Krankenhaus, wenn
- eine Lähmung schnell innerhalb von Minuten bis Stunden entstanden ist
- eine Lähmung die Harnblase oder den Afterschließmuskel betrifft
- eine Lähmung der Atemmuskulatur zu Atemnot oder Luftmangel führt.
Die Erkrankung
Krankheitsentstehung
Alle Impulse für Willkürbewegungen gehen von den ersten motorischen Nervenzellen der motorischen Großhirnrinde aus, die an der Gehirnoberfläche rechts und links des Scheitels nach unten zieht. Ihre Fortsätze leiten die Signale mitten durch das Zwischenhirn nach unten. Die Impulse für die Gesichtsmuskulatur werden im Hirnstamm, diejenigen für die übrige Skelettmuskulatur im Rückenmark auf die zweite motorische Nervenzelle umgeschaltet. Deren Fortsätze verlassen das zentrale Nervensystem und ziehen mit den Nerven zu den Muskeln, wo sie an speziellen Überträgereinheiten, den motorischen Endplatten, ihre Befehle auf den Muskel übertragen, der sich dann zusammenzieht. Ist diese Kette auch nur an einer Stelle unterbrochen, sind Lähmungen die Folge.
Formen
Eine vollständige Lähmung der Skelettmuskeln bezeichnet man als Plegie, leichtere (unvollständige) Lähmungen als Parese. Unvollständige Lähmungen werden vom Betroffenen oft als (Muskel-)Schwäche wahrgenommen. Lähmungen können nicht nur die willkürlich kontrollierbare Muskulatur, sondern auch unwillkürlich arbeitende Muskeln wie z. B. den Darm betreffen. Hier spricht man dann von häufig einer Paralyse, z. B. von einem paralytischen Ileus.
Je nach Ort der Lähmung unterscheidet man folgende Formen:
- Monoplegie: vollständige Lähmung eines Armes, eines Beines oder eines Gliedmaßenabschnitts, z. B. der Hand
- Paraplegie: vollständige Lähmung beider Beine
- Hemiplegie: vollständige Lähmung einer Körperseite und der Hälfte eines Organs, z. B. des Kehlkopfes bei halbseitiger Kehlkopflähmung
- Tetraplegie: vollständige Lähmung beider Arme und beider Beine.
Außerdem unterscheidet man die schlaffe Lähmung von der spastischen Lähmung. Die Durchtrennung eins Nervens führt zu einer schlaffen Lähmung, es ist im betroffenen Muskel keinerlei Aktivität mehr nachweisbar. Bei der spastischen Lähmung ist der Muskel spontan aktiv, wird aber nicht mehr durch das zentrale Nervensystem kontrolliert. Er hat einen hohen Eigentonus, d. h. eine hohe Eigenspannung, und die Muskelreflexe sind verstärkt auslösbar. Spastische Lähmungen sind typisch für zentrale Lähmungen.
Ursachen
Lähmungsursachen können je nach Ort der Schädigung in drei Gruppen eingeteilt werden. Zentrale Lähmungen entstehen durch Schäden im Gehirn, periphere Lähmungen durch Schäden an den außerhalb des Gehirns verlaufenden Nervenbahnen einschließlich motorischer Endplatte. Muskuläre Lähmungen beruhen dagegen auf einer Erkrankung des Muskels selbst.
- Zentrale Lähmungen. Bei der zentralen Lähmung ist die erste motorische Nervenzelle in der Großhirnrinde oder ihre Leitung (ihr Axon) zur zweiten motorischen Nervenzelle betroffen. Dies geschieht z. B. aufgrund unfallbedingter Querschnittlähmung oder Schädel-Hirn-Verletzung, Durchblutungsstörungen wie Schlaganfall und Hirntumoren oder Hirnentzündungen. Ebenfalls zu den zentralen Lähmungen gehören angeborene Hirnschäden wie z. B. bei der infantilen Zerebralparese.
- Periphere Lähmungen. Hier ist die zweite motorische Nervenzelle betroffen, und zwar entweder die Nervenzelle selbst, die im Rückenmark liegt oder ihr Axon, d. h. die Nervenfaser, die zum Muskel läuft. Hierzu gehören die Radialislähmung, idiopathische Gesichtslähmung, Lähmungen durch Rückenmarktumoren oder aufgrund von Bandscheibenvorfällen, Autoimmunerkrankungen, Guillain-Barré-Syndrom, Vergiftungen (Botulismus), Polyneuropathie oder die Schädigung der Armnerven durch die Geburtszange.
- Muskuläre Lähmungen. In diese Gruppe fallen z. B. Lähmungen durch Muskelentzündungen wie die Polymyositis und die angeborene progressive Muskeldystrophie.
Folgeschäden
Insbesondere bei ausgedehnten Lähmungen drohen Folgeschäden wie die Versteifung von Gelenken (Kontraktur) oder das Wundliegen v. a. der gelähmten Körperteile (Dekubitus). Bei Beteiligung der Rumpf- und Atemmuskulatur drohen eine Lungenentzündung und/oder eine unzureichende Atemfunktion.
Diagnosesicherung
Erster Schritt ist immer die genaue Darstellung der Beschwerden. Dazu befragt der Arzt den Patienten und zieht eventuelle Begleitsymptome mit in Betracht. Durch die sich anschließende gründliche Untersuchung kann der Arzt die Ursache oft eingrenzen. Weitere diagnostische Schritte wie MRT und CT sowie elektrische Muskelfunktionstests helfen ebenso, die Diagnose zu sichern, wie spezielle Blut- und Liquoruntersuchungen.
Differenzialdiagnosen. Abzugrenzen ist die Bewegungsunfähigkeit aus psychischen Gründen trotz intakter Muskeln und Nervenbahnen (psychisch bedingte Lähmung), der Teil eines psychiatrischen Krankheitsbilds ist. Auch Muskelverkrampfungen, schmerzbedingtes Schonen oder Bewegungsblockaden sind keine Lähmungen, auch wenn sie möglicherweise so empfunden werden.
Behandlung
Wie eine Lähmung behandelt wird und wie die Chancen auf Wiederherstellung sind, ist von der Ursache abhängig und wird daher bei den jeweiligen Erkrankungen dargestellt. Zusätzlich unterstützen entsprechende Rehabilitationsmaßnahmen. Ist beispielsweise die Sprech- und Schluckmuskulatur betroffen, kommt die Logopädie zum Einsatz.
Behandlung der spastischen Lähmung. Unabhängig von der Ursache sind bei spastischen Lähmungen Krankengymnastik, Bewegungstraining, Hydrotherapie und Schwimmen sowie therapeutisches Reiten hilfreich. Auch Tabletten können die Spastik lindern. Häufig verwendete Wirkstoffe sind Baclofen, Tizanidin und Tolperison. Nachteil der oralen Therapie mit Tabletten ist jedoch, dass sie auf alle Muskeln wirkt, dadurch zu Müdigkeit und Muskelschwäche führt und eine gleichzeitige schlaffe Lähmung zusätzlich verstärkt.
Bei schwerer Spastik spritzen die Ärzte auch Botulinumtoxin in den Muskel, um die Ausschüttung des Nerventransmitters Acetylcholin an der motorischen Endplatte zu hemmen und den Muskel dadurch vorübergehend zu entspannen. In Deutschland ist diese Behandlung für die infantile Zerebralparese und die Spastik der Hand und des Fußgelenks nach Schlaganfall zugelassen, wird aber auch off label bei anderen spastischen Lähmungen eingesetzt. Eine andere Möglichkeit, eine schwere, den ganzen Körper betreffende Spastik zu behandeln, ist die Gabe von Baclofen in den Liquor, der das Rückenmark umspült.
Prognose
Ob Lähmungen bestehen bleiben oder nur vorübergehend sind, hängt von ihrer Ursache ab. Bei manchen Lähmungen gibt es die Chance, dass sie sich wieder zurückbilden, z. B. bei der idiopathischen Gesichtslähmung, beim Schlaganfall oder nach Entfernung eines Rückenmarktumors.
Für die Aussichten ist darüber hinaus entscheidend, ob Rehabilitationsmaßnahmen greifen und z. B. nach Querschnittlähmung eventuell Restfunktionen erhalten oder sogar verstärkt werden können.
Weiterführende Informationen Webseite der Deutschsprachigen Medizinischen Gesellschaft für Paraplegiologie, die Information zur Behandlung und Rehabilitation von Menschen mit Querschnittslähmung bietet. www.dmgp.de/ Offizielle Webseite der Fördergemeinschaft der Querschnittgelähmten www.fgq.de/
Muskeldystrophien
[Progressive] Muskeldystrophien: Oberbegriff für verschiedene erbliche Muskelkrankheiten, die durch einen fortschreitenden Muskelschwund bis hin zur Lähmung gekennzeichnet sind. Bei der häufigsten Form, der Duchenne-Muskeldystrophie, sind aufgrund des X-chromosomalem Erbgangs fast ausschließlich Jungen betroffen. Sie zeigt sich früh mit Gangstörungen, ab der Pubertät sind die Kinder meist gehunfähig und benötigen einen Rollstuhl. Durch Befall der Herz- und Atemmuskulatur entwickeln sich Herzschwäche und Luftnot, die Patienten sterben oft vor dem 25. Lebensjahr. Bei den anderen Muskeldystrophien kommt es erst später zu Beschwerden und die Lebenserwartung ist weniger stark eingeschränkt.
Muskeldystrophien sind nicht heilbar. Die Therapie besteht in Behandlung der Beschwerden mit intensiver Physiotherapie, operativer Behandlung von Sehnenverkürzungen oder Wirbelsäulenverkrümmung und in späten Phasen palliativer Betreuung, z. B. mithilfe einer Heimbeatmung.
Symptome und Leitbeschwerden
- Zunehmende Muskelschwäche ohne Schmerzen
- Auffälliger Watschelgang bis zur Gehunfähigkeit
- Sprachentwicklungsstörungen
- Verdickte Waden, Spitzfüße, Hervorstehen der Schulterblätter
- Zunehmende Luftnot.
Wann zur Arztpraxis
Muskeldystrophien fallen die Kinderärzt*in oft bei einer der regelmäßigen U-Untersuchungen auf. Ansonsten ist eine Ärzt*in aufzusuchen bei
- unerklärbarer Muskelschwäche
- auffälligem Gang
- Schwäche, sich aufzurichten (das Kind stützt sich aufgrund der schwachen Oberschenkelmuskeln beim Aufrichten mit den Händen auf den Oberschenkeln ab, Gowers-Zeichen)
- Sprachentwicklungsstörungen.
Die Erkrankung
Muskeldystrophien sind erbliche Erkrankungen, die durch eine Störung des Muskelstoffwechsels zu einem fortschreitenden Muskelschwund führen. Dabei wird z. B. durch Mutationen im Dystrophin-Gen das wichtige Muskelprotein Dystrophin gar nicht oder nur vermindert gebildet. In der Folge stirbt das Muskelgewebe ab und wird zunehmend mit Fett- und Bindegewebe ersetzt.
Formen
Die verschiedenen Erkrankungen unterscheiden sich in ihrem Erbgang, Zeitpunkt und Ort des Beschwerdebeginns sowie in der Geschwindigkeit des Fortschreitens.
Am häufigsten ist die Muskeldystrophie vom Typ Duchenne, auch als aufsteigende bösartige Beckengürtelform der Muskeldystrophie bezeichnet, von der etwa vier von 100.000 Kindern betroffen sind. Aufgrund eines Gendefekts fehlt das Muskelprotein Dystrophin komplett. Der Gendefekt wird X-chromosomal vererbt, das bedeutet, dass vor allem Jungen daran leiden. Erben Mädchen das veränderte Gen, tragen sie in der Regel auf dem zweiten X-Chromosom die gesunde Variante und entwickeln keine Muskeldystrophie. Sie geben aber als sogenannte Konduktorinnen das kranke Gen an 50 % ihrer Söhne weiter, die dann – da sie kein 2. unbetroffenes X-Chromosom, sondern ein Y-Chromosom besitzen – daran erkranken. Kinder mit einer Duchenne-Muskelatrophie zeigen zunächst eine Schwäche der Becken- und Oberschenkelmuskulatur. Sie lernen spät laufen, scheinen ungeschickt und bekommen einen "watschelnden" Gang. Bald können sie z. B. Treppen nicht mehr steigen und sind schon mit etwa 13 Jahren an den Rollstuhl gefesselt, parallel dazu kommt es zu schmerzhaften Fehlstellungen von Gelenken und Knochenverformungen. Die Lähmungen breiten sich immer mehr aus und führen dadurch zu Atemstörungen mit der Gefahr einer Lungenentzündung. Durch Herzbeteiligung bildet sich eine Herzschwäche aus. Ein Teil der Betroffenen hat zusätzlich eine Intelligenzminderung, diese zeigt sich auch darin, dass manche Kinder nicht richtig sprechen lernen. Die Erkrankten werden nur selten älter als 20–25 Jahre.
Langsamer, aber prinzipiell gleich verläuft die Muskeldystrophie vom Typ Becker-Kiener oder aufsteigende gutartige Beckengürtelform der Muskeldystrophie. Auch hier ist das Dystrophin-Gen gestört, das Muskelprotein Dystrophin wird vermindert gebildet, fehlt aber nicht ganz. Die Beschwerden beginnen deshalb meist erst später, etwa mit 10 Jahren, und schreiten nur ganz langsam fort, sodass viele Erkrankte 60 Jahre und älter werden.
Außerdem gibt es Formen mit bevorzugtem Befall der Extremitäten oder des Gesichts, des Schultergürtels und der Arme.
Diagnosesicherung
Bei Verdacht erfolgt zuerst eine gründliche neurologische Untersuchung. Die Ärt*innen achtet dabei auf Lähmungen, Muskelschwäche und Veränderungen der Muskeleigenreflexe. Er prüft den Gang und lässt den Patienten sich aus der Bauchlage aufrichten (Gowers-Zeichen). Kinder mit einer Duchenne-Muskelatrophie fallen auch häufig durch Spitzfüße und durch Fetteinlagerungen verdickte Wadenmuskulatur auf.
Zur Sicherung der Diagnose und zur Kontrolle und Dokumentation des Verlaufs dienen folgende Verfahren:
- Blutuntersuchung auf die Kreatin-Phosphokinase. Dieses Enzym weist auf eine Muskelschädigung hin und ist bei der gutartigen Muskeldystrophie auf das 10–35-Fache, bei der Duchenne-Muskeldystrophie auf das 50–200-Fache erhöht.
- Elektromyografie der betroffenen Muskeln. Dabei werden feine Nadeln in einen Muskel eingestochen und darüber die elektrische Aktivität des Muskels in Ruhe und bei willkürlicher Bewegung gemessen.
- Muskelbiopsie. In manchen Fällen entnehmen die Ärzt*innen auch ein kleines Stückchen Muskelgewebe (Biopsie), um den gestörten Muskelstoffwechsel im Gewebe nachzuweisen. Dabei finden sich bei allen Muskeldystrophien abgestorbene Muskelzellen und Fett- und Bindegewebe im Muskel. Mit speziellen Färbemethoden können die Ärzt*Innen zwischen der Duchenne-Muskelatrophie und der gutartigen Becker-Kiener-Dystrophie unterscheiden.
- Molekulardiagnostik. Bei sehr hohen Werten der Kreatin-Phosphokinase veranlassen die Ärzt*innen auch statt einer Muskelbiopsie die Untersuchung einer Blutprobe auf Genmutationen. Der Nachweis von Genmutationen ist aber nicht nur für die Diagnose wichtig, sondern auch für eine genetische Beratung. Getestet werden sollten auch Familienangehörige, z. B., um herauszufinden, ob Schwestern eines Jungen mit Duchenne-Muskeldystrophie evtl. Konduktorinnen der Erkrankung sind.
- Ultraschalluntersuchung. Im Ultraschall können die Ärzt*innen umgebautes Muskelgewebe erkennen. Dieses Verfahren dient vor allem der Verlaufsbeobachtung.
- Magnetresonanztomografie. In manchen Fällen benötigen die Ärzt*innen MRT-Aufnahmen, mit denen sich der fettige Umbau der Muskeln gut erkennen lässt.
- EKG, Herz-Echo. Die Mitbeteiligung des Herzens prüfen die Ärzt*Innen, indem sie mit dem EKG und dem Langzeit-EKG nach Herzrhythmusstörungen fahnden. Eine Herzmuskelschwäche weisen sie mit dem Herz-Echo, also der Ultraschalluntersuchung des Herzens nach.
- Multispektrale optoakustische Tomografie. Mit diesem neu entwickelten, strahlungsfreien und nicht-invasiven Verfahren können die Ärzt*innen den Bindegewebsanteil im Muskel nachweisen. Die Methode ist noch in der klinischen Prüfung, sie soll in Zukunft die Dokumentation des klinischen Verlaufs verbessern und mögliche therapeutische Wirkungen nachweisen.
Differenzialdiagnosen. Fortschreitende Lähmungen mit Beginn an den Beinen weisen z. B. die spinalen Muskelatrophien, die Amyotrophe Lateralsklerose und die Polymyositis auf.
Behandlung
Bis heute können Muskeldystrophen nicht geheilt werden, die Behandlung zielt deshalb vor allem darauf, das Leiden zu lindern.
- Basismaßnahmen. Die Basismaßnahmen dienen dazu, die verbliebene Muskelfunktion zu verbessern bzw. so lange wie möglich zu erhalten. Außerdem beugen sie der Versteifung von Muskeln und Gelenken vor und verbessern Fehlhaltungen, die zu Schmerzen führen. Die Betroffenen lernen, besser mit der Erkrankung und ihren Einschränkungen umzugehen, was die Aktivitäten im täglichen Leben erleichtert und die Lebensqualität erhöht. Eine umfassende psychosoziale Betreuung hilft den Erkrankten und ihren Familien, die zunehmende Belastung auszuhalten und Entscheidungen zu treffen, z. B. bezüglich einer Heimbeatmung. In späten Stadien kommen vor allem unterstützende Maßnahmen für die Nahrungsaufnahme und die Atmung dazu. Eingesetzt werden dazu
- Physiotherapie, Krankengymnastik, Schwimmen in warmem Wasser
- Ergotherapie und Logopädie
- Atemtherapie mit Atemübungen
- Osteopathie, manuelle Therapie
- jährliche stationäre Rehabilitationen in speziellen Zentren
- Psychotherapie
- Heimbeatmung (in spätem Stadium)
- Magensonde (in spätem Stadium).
- Pharmakotherapie
- Herzmedikamente zur Unterstützung der Herzschwäche
- Ataluren (z. B. Translarna®) ist seit 2018 zugelassen für gehfähige Patienten über 2 Jahren, die an einer Duchenne-Muskeldystrophie mit speziellem Gendefekt leiden. Der Wirkstoff bessert bei einigen Patient*innen die Gehfähigkeit, allerdings leider nur in Maßen. Häufige Nebenwirkung bei dieser Therapie sind Erbrechen, Durchfall, Bauchschmerzen und Kopfschmerzen.
- Kortison soll bei der Muskeldystrophie Typ Duchenne evtl. den Krankheitsverlauf etwas verzögern, dies muss aber gegen die Nachteile einer Langzeitbehandlung abgewogen werden.
- Operationen
- Verlängerung von durch Dauerkontraktion verkürzten Sehnen
- Operative Lösung von verkürzten Muskeln
- Versteifung der Wirbelsäule zur Stabilisierung
- Genetische Beratung
- Allen Betroffenen (bzw. bei Kindern ihren Eltern) wird eine genetische Beratung angeboten, um das Wiederholungsrisiko für weitere Kinder abzuschätzen. Ein Teil der Muskeldystrophien kann bereits vor der Geburt festgestellt werden.
Prognose
Bei der häufigsten Form, der Duchenne-Muskeldystrophie, ist die Prognose ernst. Die Betroffenen sterben am fortschreitenden Abbau der Herz- und Atemmuskulatur meist im Alter von 20 bis 25 Jahren.
Die anderen Muskeldystrophien haben eine deutlich bessere Lebenserwartung als die Duchenne-Dystrophie, bei der Becker-Kiener-Variante werden viele Betroffene 60 Jahre und älter.
Ihr Apotheker empfiehlt
Die Diagnose Muskeldystrophie ist für die Betroffenen und ihre Angehörigen zunächst meist ein Schock. Auch wenn die Erkrankten in der Regel vom Zeitpunkt der Diagnose in der Kindheit an durch ein Team von Spezialisten begleitet werden, ist bei den Betroffenen und ihren Familien ein hohes Maß an Verantwortung und Selbsthilfe gefragt.
- Machen Sie sich frühzeitig Gedanken, wie Ausbildung, Berufsleben, Wohnsituation aussehen.
- Nutzen Sie alle möglichen Hilfen und Ressourcen, auf die Sie einen Anspruch haben. Zögern Sie nicht, professionelle Hilfe anzunehmen.
- Achten Sie auf Zeichen der Depression und Angst, suchen Sie frühzeitig psychotherapeutische Hilfe auf.
- Wenn Ihr Kind von einer Muskeldystrophie betroffen ist, achten Sie auf seine Sprachentwicklung und suchen Sie bei Störungen frühzeitig Hilfe.
- Bekommen Sie oder Ihr Kind Kortison verschrieben, achten Sie auf eine bewusste Ernährung, um der drohenden Gewichtszunahme vorzubeugen. Sorgen Sie für eine altersgerechte ausreichende Kalziumzufuhr.
- Wichtig ist es auch, frühzeitig darüber zu entscheiden, wie die palliative, d. h. die lindernde ärztliche und pflegerische Unterstützung aussehen soll, wenn die Muskeln von Herz oder Atemsystem immer schwächer werden. Besprechen Sie in der Familie, ob eine Heimbeatmung gewünscht ist.
- Wenn Sie selbst an einer Muskeldystrophie leiden legen Sie frühzeitig fest, wer in Notsituation bevollmächtigt ist, in Ihrem Namen Entscheidungen zu treffen.
Weiterführende Informationen
Der gemeinnützige Verein Deutsche Muskelschwund-Hilfe unterstützt Betroffene und ihre Familien mit Rat und Informationen: https://www.muskelschwund.de Einen ausführlichen Familienratgeber zur Muskeldystrophie Duchenne kann man auf der Webseite von Treat-NMD Neuromuscular Network unter folgendem Link herunterladen: https://treat-nmd.org/wp-content/uploads/2019/11/uncategorized-Duchenne_Ratgeber.pdf
Myasthenia gravis
Myasthenia gravis: Autoimmunerkrankung, die durch eine gestörte Erregungsübertragung vom Nerv auf den Muskel mit der Folge einer Muskelschwäche gekennzeichnet ist. Zu Beginn kommt es oft zu Doppelbildern, Herabhängen des Oberlids, Kau- und Schluckbeschwerden oder näselnder Sprache. Es gibt verschiedene Formen, an der (häufigsten) Variante mit spätem Krankheitsbeginn erkranken vor allem Männer, an den Varianten mit einem Krankheitsbeginn vor dem 50. Lebensjahr häufiger Frauen.
Behandelt wird die Myasthenie mit Medikamenten, die die neuromuskuläre Signalübertragung verbessern, Immunsuppression und in vielen Fällen zusätzlich operativ durch Entfernung des Thymus. Die Prognose ist heute überwiegend gut.
Symptome und Leitbeschwerden
- Unnatürlich schnelle Ermüdbarkeit der Muskulatur, die im Tagesverlauf oder unter Stress zunimmt
- Sehen von Doppelbildern
- Herabhängendes Oberlid
- Störungen beim Schlucken und Sprechen
- Schwierigkeiten beim Heben des Armes oder Schwäche beim Halten des Kopfes.
Wann zur Arztpraxis
Sofort, wenn sich bei bekannter Myasthenie-Erkrankung
- die Muskelschwäche akut verschlechtert und Atem- oder Schluckstörungen auftreten.
In den nächsten Tagen, wenn
- sich die oben genannten Beschwerden entwickeln.
Die Erkrankung
Krankheitsentstehung
Um Signale von Nerven empfangen zu können, hat der Muskel spezielle Kontaktstellen, die Acetylcholinrezeptoren, an denen der aus dem Nervenende ausgeschüttete Überträgerstoff "andockt". Bei der Myasthenia gravis bildet der Körper aus noch ungeklärter Ursache Abwehrstoffe (Autoantikörper) gegen diese Rezeptoren. Diese sogenannten Anti-Acetylcholinrezeptor-Antikörper (Anti-AChR-Antikörper) docken an die Rezeptoren an und blockieren diese. Dadurch erreicht das als Befehl zur Kontraktion ausgeschüttete Acetylcholin den Muskel nicht mehr, was zu einer schnellen Muskelermüdung bis hin zur Muskellähmung führt.
Neben den Anti-Acetylcholinrezeptor-Antikörpern gibt es weitere, gegen andere Strukturen des Muskels gerichtete Antikörper. Diese dienen zur Unterscheidung der verschiedenen Formen der Myasthenie (siehe unten). Manche Myasthenieformen werden auch als "seronegativ" bezeichnet, weil sich bei ihnen (noch) keine Autoantikörper nachweisen lassen.
Klinik und Formen
Die Erkrankung beginnt bei zwei Dritteln der Patient*innen mit Doppelbildern (durch Augenmuskelschwäche) und Herabhängen des Augenlids, oft gehören auch Schluck- und Sprechstörungen zu den ersten Beschwerden. Später kommt es je nach Form zu Schwäche von Armen und Beinen, zu einer Kopfhalteschwäche, in schweren Fällen ist auch die Atemmuskulatur betroffen und die Patient*innen entwickeln Luftnot. Typischerweise nimmt die Muskelschwäche im Verlauf des Tages und in Stresssituationen zu.
- Myasthenia gravis mit spätem Krankheitsbeginn (Late-onset Myasthenia gravis, LOMG). Diese Form betrifft vor allem Männer über 50 Jahren, sie ist mit 45 % der Myasthenie-Fälle die häufigste Variante. Die muskuläre Ermüdung ist generalisiert, d. h. sie breitet sich auf den ganzen Körper aus. Neben dem Haupt-Autoantikörper Anti-AChR-Antikörper kommen häufig weitere Antikörper gegen muskuläre Strukturen vor.
- Myasthenia gravis mit frühem Krankheitsbeginn (Early-onset Myasthenia gravis, EOMG). An dieser zu etwa 20 % auftretenden Variante erkranken vor allem Frauen unter 50 Jahren. Auch die EOMG verläuft generalisiert. Bei der EOMG lassen sich vor allem Anti-AChR-Antikörper nachweisen.
- Okuläre Myasthenia gravis (OMG, etwa 15 % der Myasthenien). Diese Form betrifft in erster Linie die Augen. Es erkranken insbesondere Frauen, und zwar in jedem Lebensalter. Bei bis zu 70 % der Patienten sind Anti-AChR-Antikörper nachweisbar.
- Thymom-assoziierte Myasthenia gravis (TAMG, etwa 15 %der Myasthenien). Diese Variante ist mit der Vergrößerung der Thymusdrüse am Hals verbunden. Es lassen sich neben dem typischen Anti-AChR-Antikörper zahlreiche weitere Antikörper nachweisen. Betroffen sind Männer und Frauen gleichermaßen, die Erkrankung beginnt zwischen dem 40. und 60. Lebensjahr und verläuft generalisiert. Bei dieser Variante entfernen die Ärzt*innen häufig den Thymus.
Anti-MuSK-AK-assoziierte Myasthenia gravis (MAMG). Seltene (5 % der Myasthenien), vor allem jüngere Frauen betreffende Variante mit dem Antikörper gegen die muskelspezifische Tyrosinkinase (einem für die Signalübermittlung an der motorischen Endplatte wichtigem Enzym). Bei dieser Form kommt es vor allem zu Augenbeschwerden (Doppelbilder, Herabhängen des Augenlids), zu Schluckstörungen und zu schlaffen Gesichtszügen aufgrund des Befalls der mimischen Muskulatur.
Diagnosesicherung
Die Diagnostik beginnt mit Belastungstests, um eine schnelle Ermüdung der Muskulatur festzustellen. Dazu dienen z. B. Armhalteversuche, bei denen die Arme nach vorn ausgestreckt gehalten werden und nach kurzer Zeit durch die Muskelschwäche absinken. Mithilfe weiterer Halteversuche des Kopfes und der Beine sowie der Prüfung des Lidhebers, der Mimik, der Kaumuskulatur und der Atmung bestimmen die Ärzt*innen außerdem den Schweregrad der Erkrankung (Besinger-Score), um den Verlauf und das Ansprechen einer Therapie dokumentieren zu können.
In Blutuntersuchungen werden manchmal Antikörper gegen den Acetylcholinrezeptor oder gegen andere Proteine nachgewiesen, um die Diagnose zu stützen.
Die Elektromyografie zeigt eine typische Abnahme der elektrischen Aktivität von Muskeln bei einer wiederholten Reizung.
Pharmakologische Tests. Beim Edrophonium-Test spritzen die Ärzt*innen probeweise intravenös das Medikaments Edrophoniumchlorid. Es bessert bei Myasthenia gravis die Symptomatik innerhalb von 60 Sekunden. Um die Wirkung dokumentieren zu können, wird die Patient*in meist vor und nach Gabe des Mittels fotografiert. Bei älteren Patient*innenen verwenden die Ärzt*innen statt Edrophonium häufig Pyridostigmin in Tablettenform. Dieser Test ist als positiv zu bewerten, wenn sich die Beschwerden des Patienten nach 45 bis 60 Minuten bessern.
Außerdem veranlasst die Ärzt*in ein CT des Brustkorbs, um nach einer Geschwulst des Thymus zu suchen, die bei der Thymom-assoziierten Myasthenia gravis vorkommt (siehe Formen der Myasthenie).
Differenzialdiagnosen. Neben der Myasthenia gravis gibt es noch einige andere Muskelerkrankungen, die zu einer Muskelschwäche führen, z. B. das Lambert-Eaton-Myasthenie-Syndrom oder andere seltene angeborene, genetisch bedingte Störungen der neuromuskulären Übertragung. Auch manche Arzneimittel lösen Muskelschwäche aus. Zu Doppelbildern und Schluckstörungen kommt es zudem beim Botulismus. Leidet die Patent*in nur unter Augenbeschwerden, muss die Ärzt*in Gehirntumoren oder Hirnarterienaneurysmen ausschließen.
Behandlung
Die Beschwerden werden bei der Mehrzahl der Patient*innen gut durch Cholinesterase-Hemmer wie Pyridostigmin (z. B. Mestinon®) gelindert, welche den Abbau des Überträgerstoffs Acetylcholin verringern und dadurch dessen Konzentration an den Kontaktstellen erhöhen. Der Neurologe oder die Neurologin verordnet das Medikament einschleichend und passt die Dosis dem therapeutischen Effekt an. Da die Wirkung nur etwa 3–6 Stunden anhält muss die Patent*innen die Tabletten 3–4 Mal am Tag einnehmen. Häufige unerwünschte Wirkungen sind Übelkeit, Bauchkrämpfe, Durchfall, verlangsamter Herzschlag und vermehrtes Schwitzen. In der Regel lassen die Nebenwirkungen im Verlauf der Therapie nach.
Reichen Cholinesterase-Hemmer nicht aus, bekommt die Patent*innen zusätzlich Immunsuppressiva, um die fehlgeleiteten Abwehrvorgänge im Körper zu dämpfen. Dabei verordnet die Ärzt*in Kortison, Azathioprin (z. B. Imurek®) oder ein anderes Immunsuppressivum (Rituximab, Cyclosporin A, Tacrolimus), entweder als Monotherapie oder miteinander kombiniert. Bei allen Immunsuppressiva muss regelmäßig das Blutbild kontrolliert werden, da sie die weißen Blutzellen, die Erythrozyten und die Thrombozyten verringern können. Bei der regelmäßigen Einnahme von Kortison sind außerdem auf die durch diesen Wirkstoff häufigen Nebenwirkungen zu achten, wie beispielsweise Knochenschwund (Osteoporose), Gewichtszunahme, Linsentrübung und Störungen der Sexualfunktion.
Thymektomie. Besteht eine Geschwulst des Thymus, wird der Thymus operativ entfernt. In einigen Fällen empfehlen die Ärzt*innen die Operation auch, wenn der Thymus nicht vergrößert ist. So zum Beispiel, wenn die medikamentöse Therapie keine Wirkung zeigt oder bei Patient*innen mit einer generalisierten Myasthenie.
Bei sehr schweren Formen oder bei der myasthenen Krise versuchen die Ärzte, die schädigenden Antikörper mithilfe einer Plasmapherese (Blutwäsche) zu entfernen. Eine andere Methode ist die Gabe von hochdosierten Immunglobulinen über mehrere Tage, wobei das Wirkprinzip dieser Off-Label-Behandlung unklar ist.
Prognose
Über 90 % der Betroffenen können mit den Medikamenten weitgehend normal leben, berufstätig sein und sogar Sport treiben. Berufe mit körperlicher Dauerbelastung sind allerdings nicht möglich.
Von den Patient*innen, die eine myasthene Krise entwickeln, sterben trotz Behandlung 2–3 %. Unbehandelt ist die Letalität deutlich höher.
Ihre Apotheke empfiehlt
- Achten Sie bei Infekten und Fieber gut auf Ihre muskulären Beschwerden. Sollten Sie Schluck- oder Atemstörungen entwickeln, suchen Sie unverzüglich Ihre Ärzt*in auf.
- Halten Sie sich genauestens an die vom Arzt oder Ärztin verschriebene Dosierung und Einnahmehinweise Ihrer Myasthenie-Medikamente. Eine Über- oder Unterdosierung kann schwerwiegende Folgen wie z. B. eine myasthene Krise haben.
- Tragen Sie immer Ihren Myastheniepass bei sich, damit z. B. bei einem Unfall keine falschen Medikamente gegeben werden.
- Schließen Sie sich einer Selbsthilfegruppe an oder suchen Sie anderweitig Informationen und Rat (Links zu informativen Webseiten siehe "Weiterführende Informationen").
- Bei Sprechstörungen hilft die logopädische Behandlung, Doppelbilder lassen sich mit speziellen Brillen lindern. Je nach Ausmaß der Erkrankung haben Sie Anspruch auf weitere Hilfsmittel, z. B. eine Halskrause zur Kopfunterstützung, Gehstock, Rollator oder Rollstuhl.
- Sprechen Sie bei Kinderwunsch mit Ihrer Ärzt*in, da eine Medikamentenumstellung erforderlich sein kann.
Weiterführende Informationen
Die deutsche Myasthenie Gesellschaft e.V. bietet Betroffenen Hilfe, Unterstützung und Austausch unter https://dmg-online.de/ Informationen für Betroffene und Angehörige finden sich auf https://www.myasthenia-gravis.eu/ Über die Webseite der Deutschen Gesellschaft für Muskelkranke e.V. kann man Kontakt zu Diagnosegruppen bekommen: https://www.dgm.org/diagnosegruppen
Polyneuropathie
Polyneuropathie (PNP, Polyneuritis): Nicht verletzungsbedingte Funktionsstörungen mehrerer peripherer Nerven, die sich oft durch Ameisenlaufen, Kribbeln oder Schmerzen äußern. Zu den häufigsten Ursachen gehören zu je einem Drittel der Fälle Diabetes und Alkoholabhängigkeit, allerdings können auch Infektionen, Vergiftungen, Gefäßentzündungen oder angeborene Erkrankungen zu Polyneuropathien führen.
Neben der Behandlung der zugrundeliegenden Erkrankung kommen Schmerzmittel, Antiepileptika und Antidepressiva zum Einsatz. Meist ist damit jedoch nur eine Linderung der Beschwerden und keine völlige Schmerzfreiheit zu erzielen.
Symptome und Leitbeschwerden
- Symmetrische, distal betonte, strumpf- oder handschuhförmige schmerzhafte oder brennende Missempfindungen
- "Ameisenlaufen" oder Kribbeln auf der Haut
- "Burning feet", d. h. Brennen an den Füßen, vor allem nachts
- Wadenkrämpfe
- Vermindertes Berührungs- oder Vibrationsempfinden, Taubheitsgefühle oder auch eingeschränktes Schmerzempfinden
- Gehstörungen
- Lähmungen
- Blasenschwäche, Erektionsstörungen, Völlegefühl.
Wann in die Arztpraxis
In den nächsten Tagen, wenn
- oben genannte Missempfindungen oder Empfindungsstörungen über mehrere Tage bestehen oder sich schnell verstärken.
Noch am selben Tag
- bei Muskellähmungen.
Die Erkrankung
Verlauf
Polyneuropathien beginnen in der Regel schleichend, häufig mit strumpf- oder handschuhförmigen Empfindungsstörungen an Beinen bzw. Armen. Während in erster Linie Missempfindungen und Schmerzen den Betroffenen belasten und daher zur Arztpraxis führen, werden Ausfälle wie etwa ein vermindertes Vibrationsempfinden erst spät bemerkt, etwa wenn es durch fehlende Rückmeldung über die Bodenbeschaffenheit zu Gehstörungen gekommen ist.
Sind Nerven beeinträchtigt, die für Muskelbewegungen zuständig sind, kommt es zu Muskelschwäche oder Lähmungen. Sind Nerven des vegetativen Nervensystems betroffen, können Magen-Darm-Beschwerden, sexuelle Störungen und Blasen- oder Darmentleerungsstörungen folgen. Führt die Nervenschädigung zu einer gestörten Blutdruckregulation, entwickelt sich manchmal eine orthostatische Dysregulation mit Schwindelgefühl beim Aufstehen aus dem Liegen. Auch die Hautdurchblutung leidet, was sich z. B. durch hartnäckigen Hautpilz der Zehen zeigen kann, nicht selten aber erst bei Vorliegen von Hautgeschwüren bemerkt wird.
Ursachen
Unzählige Erkrankungen und Substanzen können die Nerven schädigen. Zu den wichtigsten Ursachen gehören
- Toxische Einflüsse
- Alkoholmissbrauch
- Vergiftungen mit Schwermetallen wie Blei oder Arsen
- Lösungsmittel
- Medikamente (Chemotherapeutika)
- Stoffwechsel- oder hormonelle Erkrankungen
- Diabetes mellitus
- Akromegalie
- Schwangerschaft
- Mangelernährung (z. B. Vitamin-B-Mangel)
- Angeborene Erkrankungen
- Porphyrie
- Amyloidose
- Genetisch bedingte Neuropathien (selten)
- Infektionen
- Borreliose, Lepra, Syphilis
- HIV-Infektion, Zytomegalie, Influenza
- Entzündungen
- rheumatische Erkrankungen
- Guillain-Barré-Syndrom
- Therapeutische Bestrahlung, z. B. im Rahmen einer Krebsbehandlung
- Gefäßentzündungen
- Begleiterscheinung einer Krebserkrankung
- Z. B. beim Lungenkrebs.
Diagnosesicherung
Die Ursacheneingrenzung gleicht manchmal der berühmten Suche nach der Nadel im Heuhaufen (und bleibt in etwa 20 % erfolglos). Sie ist aber notwendig, weil die Polyneuropathie bei weiter bestehender Schädigungsursache fortschreitet, wohingegen die Chancen auf (langsame) Besserung der Beschwerden nach Beseitigung der Ursache gut sind.
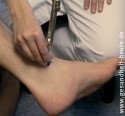
Die Diagnostik beginnt mit diversen neurologischen Untersuchungen, wobei die Ärzt*in Sensibilität, Vibrationsempfinden (siehe Abbildung), Motorik und Reflexe genau prüft. Außerdem folgen Blutuntersuchungen auf mögliche Auslöser, z. B. auf Giftstoffe, erhöhten Blutzucker, Syphilis-Antikörper oder Vitaminmangel.
Erhärtet wird der Verdacht auf eine Polyneuropathie durch eine Messung der Nervenleitgeschwindigkeit und eine Elektromyografie. Beide Untersuchungen sind unangenehm, da feine Nadeln unter die Haut eingestochen werden, aber gut erträglich.
Im Zweifel werden auch kleine Gewebeproben entnommen, z. B. von Nerven oder von der Haut, um diese unter dem Mikroskop untersuchen zu lassen.
Ob die autonomen Nervenfasern des Herzens geschädigt sind, wird mithilfe des EKGs geprüft. Eine Schädigung der Blasennerven äußert sich zumeist durch die inkomplette Blasenentleerung der Blase, d. h., es verbleibt nach dem Wasserlassen Restharn in der Blase. Diesen Restharn wird z. B. mithilfe einer Ultraschalluntersuchung nachgewiesen.
Differenzialdiagnosen. Symmetrisch verteilte Missempfindungen und Schmerzen an Armen oder Beinen finden sich auch bei der Peripheren arteriellen Verschlusskrankheit (pAVK), Funikulärer Myelose, Neurosyphilis oder bei Nervenkompression durch Bandscheibenvorfälle.
Behandlung
Neben der Ursachenbehandlung (Diabetes optimal einstellen, Alkoholkonsum reduzieren, Infektionen behandeln) verordnet die Ärzt*in zusätzlich Medikamente gegen die oft quälenden Missempfindungen und Schmerzen. Dabei ist Geduld gefragt, da die Medikamente oft mehrere Wochen brauchen, um wirksam zu werden. Zudem lässt sich meist keine völlige Schmerzfreiheit erzielen, häufig ist jedoch eine Linderung der Schmerzen auf ein akzeptables Niveau möglich.
Bei leichten, nicht kontinuierlichen Schmerzen empfehlen die Leitlinien einen Behandlungsversuch mit Schmerzmitteln wie Paracetamol oder Metamizol. Reichen diese nicht aus oder handelt es sich von vorneherein um mittelschwere oder starke Schmerzen und Missempfindungen, werden folgenden Wirkstoffe einzeln oder in Kombination verabreicht:
- Antiepileptika wie z. B. Gabapentin, Pregabalin, Carbamazepin. Diese krampflösenden Substanzen dämpfen die Erregbarkeit der Nervenzellen und können dadurch Schmerzen und Missempfindungen lindern. Antiepileptika stören oft die Blutbildung, deshalb muss die Ärzt*in regelmäßig das Blut kontrollieren.
- Antidepressiva wie z. B. Amitryptilin oder Duloxetin. Sie beseitigen den Schmerz nicht, unterdrücken aber seine Weiterleitung. Antidepressiva dosiert der Arzt vorsichtig auf, um die Nebenwirkungen wie Blutdruckabfall oder Erektionsstörungen möglichst gering zu halten.
- Opioide sind starke Schmerzmittel, haben aber den Nachteil, dass sie zu einer Gewöhnung führen. Manchmal lässt auch ihre Wirkung mit der Zeit nach, wodurch immer höhere Dosen erforderlich sind (Toleranzentwicklung). Sie werden nur verordnet, wenn Antiepileptika oder Antidepressiva keine Wirkung zeigen.
- Schmerzlindernde Pflaster. Schmerzlindernde Pflaster gelten in der Therapie des polyneuropathischen Schmerzes als zweite Wahl, d. h. die Ärzt*in verordnet sie, wenn die Therapie mit Tabletten nicht anschlägt. Sie dürfen nur auf unverletzte, trockene Haut geklebt werden. Capsaicin-Pflaster (z. B. Qutenza®) ist zugelassen zur Behandlung polyneuropathischer Schmerzen bei Erwachsenen, die nicht an Diabetes leiden. Der Wirkstoff reduziert die Empfindlichkeit der Schmerzrezeptoren in der Haut. Etwa eine Woche, nachdem das Capsaicin-Pflaster für 30–60 Minuten auf das schmerzende Areal aufgeklebt wurde, beginnt die Schmerzreduktion, die über etwa 12 Wochen anhält. Lidocain-Pflaster unterbinden die krankhafte Nervenerregung in der Haut, sie werden einmal täglich für 12 Stunden auf die Haut geklebt.
- Alpha-Liponsäure soll Schmerzen und Empfindungsstörungen bei der diabetischen Polyneuropathie verbessern, die Wirkung ist allerdings umstritten. Deshalb übernehmen die Gesetzlichen Krankenkassen die Kosten einer solchen Behandlung nicht.
Physio- und Ergotherapie helfen, Gelenkversteifungen zu vermeiden und Muskeln wiederaufzubauen. Individuell angepasste Hilfsmittel können die Gangsicherheit erhöhen und Immobilität vorbeugen.
Prognose
Lässt sich bei einer Polyneuropathie der Auslöser frühzeitig abstellen, ist die Prognose gut. Je später jedoch die Erkrankung erkannt wird, desto höher ist die Gefahr, dass die Nerven schon bleibend geschädigt sind. In diesen Fällen lassen sich die Beschwerden mit den genannten Medikamenten jedoch häufig zumindest lindern.
Ihre Apotheke empfiehlt
Was Sie selbst tun können
Füße gut pflegen. Die Empfindungsstörungen bei Polyneuropathien können gefährliche Folgen haben. Dadurch, dass Druckstellen oder kleine Fußverletzungen nicht mehr bemerkt werden, entwickeln sich sehr leicht Fußgeschwüre. Vor allem Diabetiker sind häufig vom diabetischen Fußsyndrom betroffen, weil bei ihnen zusätzlich die Wundheilung gestört ist. Achten Sie deshalb auf eine gute Fußpflege und tragen Sie nur gut passendes Schuhwerk.
Mobil bleiben. Nutzen Sie physikalische Therapien und Krankengymnastik, um gelenkig zu bleiben. Auch Wechselbäder können die Durchblutung steigern und schwache Muskeln wieder stärken.
Wadenkrämpfe lindern. Zur Behandlung von Wadenkrämpfen kann Magnesium versucht werden. In manchen Fällen verschreibt die Ärzt*in auch das rezeptpflichtige Chinin.
Schwindel- und Schwächegefühle behandeln. Wenn die Blutdruckregulation gestört ist und Sie unter einer orthostatischen Dysregulation leiden, tragen Sie Stützstrümpfe. Außerdem sollten Sie immer langsam aufstehen, um keinen Schwindel zu provozieren. Regelmäßiges Muskeltraining fördert zudem die Venenpumpe.
Blasenstörungen in den Griff bekommen. Gehen Sie regelmäßig zur Toilette, damit sich nicht zu viel Restharn in der Blase ansammelt. Suchen Sie bei starker Beeinträchtigung ihre Hausarztpraxis auf, diese kann Ihnen mit Medikamenten helfen.
Erektionsstörungen ansprechen. Polyneuropathien machen auch vor dem vegetativen Nervensystem nicht halt. Zudem stören auch die zur Behandlung der Missempfindungen eingenommenen Medikamente manchmal die Erektion. Sprechen Sie bei Erektionsstörungen mit Ihrer Ärzt*in, er kann Ihnen beispielsweise mit der Verordnung eines PDE-5-Hemmers oder auch einer Vakuumpumpe helfen.
Komplementärmedizin
Magnettherapie. Neuere Studien zeigen positive Effekte bei diabetischer Neuropathie. Auf welchem Mechanismus die Wirkung der Magnettherapie beruht, ist aber derzeit noch unklar.
Transkutane elektrische Nervenstimulation (TENS). Manchen Patienten hilft es, eine schmerzhafte Hautregion elektrisch zu stimulieren. Ob die TENS bei Polyneuropathie wirksam ist, wurde bisher jedoch nicht in kontrollierten Studien nachgewiesen.
Homöopathie. Die Homöopathie empfiehlt u. a. Aconitum C3, D4 bei neuralgischen, stechenden, brennenden Schmerzen, Agaricus muscarius D6 und D12 bei Missempfindungen (z. B. Taubheitsgefühl und "Ameisenlaufen"), Spigelia D6 und D12 bei periodisch auftretenden neuralgischen Schmerzen und Verbascum D1, D2 und D3 bei neuralgischen Gliederschmerzen mit Lähmungsgefühl für den Akutfall, sowie individuell abgestimmte Mittel zur Konstitutionstherapie.
Weiterführende Informationen
Die Deutsche Polyneuropathie Selbsthilfe e.V. unterstützt Betroffene und ihre Familienangehörigen. Die Webseite findet sich unter https://polyneuro.de/index.php?id=16
Seite mit Blogs zum Leben mit Polyneuropathie von Betroffenen: https://polyneuropathie-pnp.jimdofree.com/.
Ein Faltblatt mit Informationen zur Polyneuropathie finden Sie auf der Webseite der Deutschen Gesellschaft für Muskelkranke e.V.: https://www.dgm.org/muskelerkrankungen/polyneuropathie
Restless-legs-Syndrom
Restless-legs-Syndrom (RLS, Syndrom der unruhigen Beine): Vor allem in Ruhephasen (z. B. nachts) auftretende Missempfindungen und Unruhegefühl in den Beinen mit heftigem Bewegungsdrang oft unklarer Ursache. Betroffen sind 5–10 % der Bevölkerung, Frauen häufiger als Männer. Meist beginnt die chronische Erkrankung nach dem 30. Lebensjahr.
Leitbeschwerden
- Schwer zu unterdrückender, tiefsitzender Bewegungsdrang der Beine, in der Regel gekoppelt an unangenehme Empfindungsstörungen (z. B. Kribbeln) tief in den Beinen, meist von den Unter- zu den Oberschenkeln aufsteigend
- Möglicherweise zusätzlich gleichartige Beschwerden der Arme
- Zunahme der Beschwerden bei Ruhe und Entspannung
- Besserung der Beschwerden durch Bewegung
- Tagesrhythmik der Beschwerden mit Verstärkung am Abend und in der Nacht (vor allem beim Zubettgehen), dadurch Einschlafprobleme.
Die Erkrankung
In der Hälfte der Fälle lässt sich keine Ursache für die Erkrankung finden. Ein solches idiopathisches Restless-legs-Syndrom ist vermutlich erblich bedingt (mit mehreren betroffenen Familienmitgliedern), wobei eine Störung im Botenstoffwechsel des Gehirns (genauer im Dopaminstoffwechsel) vermutet wird. Beim symptomatischen (sekundären) Restless-legs-Syndrom dagegen lässt sich eine Ursache feststellen, am häufigsten ein Eisen-, Vitamin-B12- oder Folsäuremangel, eine Nierenfunktionsstörung oder eine rheumatische Gelenkerkrankung.
Das macht der Arzt
Die Diagnose des Restless-legs-Syndroms wird anhand des Beschwerdebildes gestellt. Blutuntersuchungen sollen behandelbare Ursachen aufdecken und die Messung der Nervenleitgeschwindigkeit eine Polyneuropathie ausschließen, die ähnliche Beschwerden bereiten kann, aber anders behandelt wird. Der Aktigraf dient der objektivierbaren Aufzeichnung der Beinbewegungen.
Besteht ein Eisen-, Vitamin-B12- oder Folsäuremangel, lassen sich die Beschwerden häufig durch eine Substitution der Nährstoffe lindern. Schwieriger ist die Behandlung beim idiopathischen Restless-legs-Syndrom. Sind die Schlafstörungen so stark, dass der Betroffene tagsüber durch Müdigkeit sehr eingeschränkt ist, helfen Medikamente. Mittel der Wahl sind das auch bei der Parkinson-Behandlung eingesetzte L-Dopa und die Dopaminagonisten, was die Hypothese von der Botenstoffwechselstörung als Ursache stützt. Bei unzureichendem Erfolg können Opiate oder bestimmte Antiepileptika versucht werden. Derzeit gibt es in Deutschland ein Opiat mit Zulassung zur Behandlung schwerer bis sehr schwerer Formen des Restless-legs-Syndroms: Targin®. Es enthält eine Kombination aus den Wirkstoffen Oxycodon und Naloxon und wird in einer Dosierung eingesetzt, die unterhalb der in der Schmerztherapie üblichen Opiatdosis liegt. Erste Studien legen eine gute Wirksamkeit nahe. Häufige Nebenwirkungen sind Kopfschmerzen, Müdigkeit, Übelkeit und Verstopfung.
Selbsthilfe
Nicht wenige Betroffene haben ihre eigenen „Rezepte“ entwickelt, um die Beschwerden in Grenzen zu halten, etwa abendliches Fahrradfahren, Schwimmen, Bürsten- oder andere Massagen der Beine. Solange der Patient mit seinen Beschwerden leben kann, ist keine Behandlung erforderlich.
Diskutiert wird ein Zusammenhang zwischen Restless-legs-Syndrom und Übergewicht, da übergewichtige Menschen signifikant häufiger ein Restless-legs-Syndrom entwickeln als schlanke. Ob eine Gewichtsabnahme die Beschwerden lindert, ist allerdings unklar.
Komplementärmedizin
- Es liegen positive Berichte der Homöopathie über eine individuelle Konstitutionsbehandlung mit Hochpotenzen vor, zur Selbstbehandlung wird Zincum valerianicum D4 empfohlen.
- Orthomolekularmedizin. Für die zusätzliche Einnahme von Vitamin- (z. B. Vitamin E oder Vitamine aus der B-Familie) oder Magnesiumpräparaten, wie mitunter empfohlen wird, sollten Sie sich nur dann entscheiden, wenn es Ihnen nicht möglich ist, den täglichen Körperbedarf dieser Nährstoffe durch die Nahrung zu decken.
Zur Linderung von Einschlafstörungen.
Weiterführende Informationen
- www.restless-legs.org – Website der Deutschen Restless-Legs-Vereinigung e. V. (RLS, München): Bietet Hintergrundinformationen, Diskussionsforen sowie eine hilfreiche Literatur- und Linksammlung.
Schwindel
Schwindel (Vertigo): Gleichgewichtsstörung, bei der der Betroffene fälschlicherweise meint, die Umgebung bewege sich, oft verbunden mit Übelkeit und Erbrechen. Schwindel ist keine eigenständige Krankheitsdiagnose, sondern ein Symptom, das bei vielen unterschiedlichen Erkrankungen und Störungen vorkommt. Häufige Ursachen sind Innenohrerkrankungen, Gefäßerkrankungen, neurologische und psychische Störungen oder unerwünschte Medikamentenwirkungen. Beim multimodalen Altersschwindel kommen meist mehrere Schwindelursachen zusammen.
Die Behandlung des Schwindels richtet sich nach der Ursache, z. B. genügt beim benignen paroxysmalen Lagerungsschwindel ein sog. Befreiungsmanöver (ein Positionswechsel nach festem Muster), während beim Akustikusneurinom operiert werden muss. Zur Akuttherapie und in Fällen, in denen sich keine Ursache finden lässt, verordnet der Arzt auch Medikamente gegen Schwindel, die Antivertiginosa. Beim multimodalen Altersschwindel sind zudem Gangschulung und Gleichgewichtstraining von besonderer Bedeutung.
Symptome und Leitbeschwerden
- Schwindelgefühle, Gefühle "wie im Karussell", "wie auf einem Schiff" oder "wie in einem auf- und abfahrenden Lift"
- Benommenheit, schummriges Gefühl im Kopf
- Gangunsicherheit
- Übelkeit und Erbrechen
- Evtl. begleitende Hörstörungen und/oder Kopfschmerzen.
Wann zum Arzt
Sofort zum Arzt oder ins Krankenhaus, bei
- plötzlich auftretendem, starkem und unerklärlichem Schwindel
- plötzlich aufgetretenen Gangstörungen, Benommenheit
- plötzlichen Seh- und Hörstörungen.
Demnächst, wenn
- sich Gangunsicherheiten oder Gleichgewichtsstörungen entwickeln
- immer wieder Benommenheit oder Schwindel auftritt
- Sehen und Hören kontinuierlich schlechter wird.
Die Erkrankung
Schwindel gehört zu den Gesundheitsproblemen, die zwar nicht altersbedingt sind, aber mit zunehmendem Alter häufiger werden: Klagen schätzungsweise 10 % aller Patienten einer Hausarztpraxis über Schwindel, so sind es bei den über 65-Jährigen bis zu 30 % und bei hochbetagten Pflegeheimbewohnern sogar bis zu 70 %.
Krankheitsentstehung
Augen, Gleichgewichtsorgane und verschiedene Reizaufnehmer (Rezeptoren) in Muskeln, Gelenken und Haut leiten ständig Informationen über unsere Position im Raum zum Gehirn weiter. Schwindel entsteht immer dann, wenn sich diese Informationen widersprechen, sodass das Gehirn kein stimmiges Bild erstellen kann. Begleitet wird Schwindel häufig von Übelkeit bis hin zu Erbrechen, Blässe und Schweißausbruch, da die an der Gleichgewichtsregulation beteiligten Nervenzellen im Gehirn mit denen zur Steuerung vegetativer Reaktionen verbunden sind.
Schwindel als solcher ist kein krankhaftes Phänomen. Jeder kennt das Kinderspiel, erst länger im Kreis gedreht und dann losgelassen zu werden. Schwindel, scheinbare Drehbewegungen der Umwelt und Taumeln sind dabei absolut normal und machen den Reiz des Spiels aus. Schwindel bei einfachem Drehen des Kopfs hingegen ist ein Krankheitszeichen.
Schwindelformen
Für die Ursachenfindung ist eine genaue Beschreibung des Schwindels unverzichtbar, wobei folgende Schwindelformen unterschieden werden:
- Zum systematischen Schwindel oder gerichteten Schwindel gehören
- Drehschwindel. Der Betroffene hat das Gefühl, seine Umgebung drehe sich um ihn wie in einem Karussell
- Schwankschwindel. Hier hat man das Gefühl, als ob der Boden unter den Füßen schwanke, etwa wie auf einem Boot
- Liftschwindel. Hier meint der Betroffene, sich wie in einem Aufzug nach oben oder unten zu bewegen
- Der ungerichtete, unsystematische oder auch diffuse Schwindel wird auch Benommenheitsschwindel genannt und geht mit dem Gefühl der "Schummrigkeit" oder "Duseligkeit" sowie häufig mit Unsicherheit bzw. Gangunsicherheit einher.
Ursachen
Die Ursache für Schwindel kann in allen an der Reizaufnahme und -verarbeitung beteiligten Strukturen liegen. Deshalb wird Schwindel durch eine große Anzahl von Erkrankungen, Störungen oder Situationen ausgelöst. Der Arzt unterscheidet neben dem "normalen" physiologischen Schwindel aufgrund von Höhe, Karussellfahren oder Reisekrankheit zwei Hauptgruppen:
Beim vestibulären Schwindel liegt die Ursache in Gleichgewichtsorgan, Kleinhirn oder Stammhirn. Hier dominiert der Drehschwindel als Beschwerde, häufig kommen aber auch Schwank- und Liftschwindel und manchmal auch Benommenheitsschwindel vor. Mögliche Ursachen für vestibulären Schwindel sind
- Gutartiger plötzlich auftretenden Lagerungsschwindel (benigner paroxysmaler Lagerungsschwindel, BPPV). Hier handelt es sich um eine der häufigsten Schwindelformen mit sekundenlangen Drehschwindelattacken, Übelkeit und einer kurz dauernden Augenbewegungsstörung. Ihm liegt eine Ablösung von winzigen Steinchen aus dem Innenohr zugrunde, die meist in den hinteren Bogengang fallen und bei schnellen Kopfbewegungen den Schwindel auslösen.
- Bilaterale Vestibulopathie. Bei dieser Erkrankung des Gleichgewichtsorgans kommt es zu bewegungsabhängigem Schwankschwindel und Gangunsicherheit, verstärkt in Dunkelheit und auf unebenem Grund.
- Morbus Menière. Hier sind anfallsartiger Drehschwindel mit Übelkeit und Erbrechen, Hörminderung und Tinnitus die typischen Beschwerden.
- Neuritis vestibularis. Wahrscheinlich handelt es sich dabei um eine durch Infektion ausgelöste Entzündung des Gleichgewichtsnervs, die zu einem Dauerdrehschwindel mit Übelkeit, Erbrechen und Fallneigung führt. Je nach Schwere der Entzündung halten sich die Symptome bis zu drei Wochen. In der Regel heilt eine Neuritis vestibularis folgenlos ab.
- Vestibuläre Migräne. Es handelt sich um eine Migräneform mit Dreh- und Schwankschwindel.
- Hirnstamm- oder Kleinhirnläsionen. Hier kann es zu Schwindel in Form von Dreh- oder Schwankschwindel und/oder Gangstörungen, Störungen der Bewegungskoordination und Sprachstörungen kommen. Ursachen sind beispielsweise Schlaganfälle oder Tumore.
- TIAs (tranistorische ischämische Attacke), also kurzfristige Durchblutungsstörungen im Kleinhirn. Hier sind Schwankschwindelattacken für Minuten bis Stunden typisch.
- Vergiftung mit Alkohol oder Drogen. Dabei kommt es meist zu ungerichtetem Benommenheitsschwindel, manchmal auch zu Drehschwindel.
- Akustikusneurinom. Hier sind neben dem Schwindel die einseitige Schwerhörigkeit und Tinnitus typisch.
- Ototoxische Medikamente. Hierbei handelt es sich um Wirkstoffe, die das Innenohr schädigen und Hörstörungen, aber auch Schwindel auslösen können. Mögliche Präparate sind Furosemid, Chinin oder Gentamycin.
Der nicht-vestibuläre Schwindel oder diffuse Schwindel hat seinen Auslöser außerhalb des Gleichgewichtsorgans. Hier leiden die Betroffenen vor allem unter dem Benommenheitsschwindel, Mischformen mit gerichteten Schwindeltypen sind jedoch auch möglich. Mögliche Ursachen sind:
- Schlechtes Sehen durch Fehlsichtigkeit oder Augenbewegungsstörungen
- Multimodaler Altersschwindel. Dieser im Alter sehr häufige Schwindel kann nicht auf eine einzelne Ursache zurückgeführt werden, sondern ist durch ein Zusammenspiel verschiedener ungünstiger Einflüsse bedingt, z. B. schwankender Blutdruck plus schlechtes Sehen plus Nervenschäden (z. B. bei Zuckerkrankheit) plus depressive Verstimmung plus eine Vielzahl eingenommener Medikamente. Typisch sind ungerichteter Schwindel, Gangunsicherheit und Schwankschwindel
- Nervenschäden mit beeinträchtigter Tastempfindung in der Haut, wodurch keine oder unpassende Meldungen an das Gehirn weitergeleitet werden, z. B. bei der Polyneuropathie
- Blutdruckabfall bei gestörter Kreislaufregulation oder Herzrhythmusstörungen, typisch ist der Benommenheitsschwindel
- Bluthochdruck (arterielle Hypertonie), vor allem wenn der Blutdruck rasch und stark ansteigt (Hypertensive Krise), aber auch beim schon länger bestehenden, moderaten Bluthochdruck.
- Subclavian-Steal-Syndrom. Hier kommt es durch Gefäßverengung der A. subclavia zu Minderdurchblutung des Gehirns bei körperlicher Belastung des Armes. Beschwerden sind u. a. plötzlicher Schwindel, Synkopen und Sehstörungen
- Funktionelles HWS-Syndrom nach einer Verletzung
- (Überdosierte) Medikamente gegen zu hohen Blutdruck oder Epilepsie sowie Schlaf- und Beruhigungsmittel. Sie führen zu Müdigkeit, Benommenheit und damit auch Schwindel
- Erkrankungen des Gehirns und der Hirnnerven wie z.B. Durchblutungsstörungen (insbesondere TIA und Schlaganfall, hier ist der Schwindel stark und akut auftretend), intrakranielle Hypertension und Schädigungen des Sehnervs.
Weitere Schwindelursachen. Eine häufige Erkrankung ist der phobische Schwankschwindel, bei dem eine angstbesetzte Auslösesituation für die Beschwerden verantwortlich ist. Physiologisch, aber äußerst unangenehm sind Höhenschwindel und der Schwindel bei Reisekrankheit, auch Kinetose genannt. Bei den beiden letztgenannten Schwindeln variieren die Beschwerden, es können Benommenheitsschwindel, Drehschwindel und Schwankschwindel auftreten.
Folgen des Schwindels
Durch die schwindelbedingte Stand- und Gangunsicherheit sind vor allem ältere Menschen erheblich gefährdet, zu stürzen und einen Knochenbruch zu erleiden. Gleichzeitig neigen sie dazu, aus Angst vor einem Sturz ihre Aktivitäten auf ein Minimum zu reduzieren, was langfristig zur Immobilität mit all ihren Folgeschäden führt.
Diagnosesicherung
Mithilfe einer gründlichen Befragung grenzt der Arzt die möglichen Ursachen des Schwindels ein. Dabei kommt es nicht nur auf Art und Dauer des Schwindels an, sondern auch darauf, ob eventuelle Begleitbeschwerden bestehen. Ein "Schwarzwerden vor den Augen" deutet z. B. auf eine kreislaufbedingte Ursache hin, Kopfschmerzen auf die vestibuläre Migräne. Wichtig ist es auch, mögliche Auslöser wie Medikamente oder Situationen zu erkennen. Die weiteren diagnostischen Maßnahmen hängen von der vermuteten Ursache ab, ein typisches Programm umfasst z. B.
- eine neurologische und HNO-ärztliche Untersuchung (letztere besonders bei Drehschwindel)
- einen Schellong-Test (zum Ausschluss oder Nachweis einer Blutdruckregulationsstörung)
- ein Langzeit-EKG (zum Ausschluss von Herzrhythmusstörungen)
- eine Dopplersonografie der Hirnarterien (um Verengungen als Ursache kurzzeitiger Unterbrechungen der Gehirndurchblutung auszuschließen)
- gegebenenfalls ein MRT (zum Ausschluss eines Schlaganfalls oder Tumors)
- sowie die Untersuchung des Liquors und verschiedene Bluttests.
Differenzialdiagnosen. Bei der Beschwerde "Schwindel" kommen alle oben genannten Störungen und Erkrankungen in Frage.
Behandlung
Die Behandlung des Schwindels ist ursachenabhängig. Feststellbare Grunderkrankungen wie etwa Herzrhythmusstörungen werden immer entsprechend therapiert.
Konservative Behandlung
Zur Behandlung des benignen paroxysmalen Lagerungsschwindels gibt es zwei sogenannte Befreiungsmanöver (nach Epley und nach Semont). Dabei werden die schwindelverursachenden Steinchen durch koordinierte Bewegungen von Kopf und Körper und durch die Schwerkraft aus den Bogengängen herausmanövriert. Links zu anschaulichen Videos siehe unter "Weiterführende Informationen".
In seiner Wirkung vielfach unterschätzt wird das Gang- und Gleichgewichtstraining. Beim gutartigen plötzlich auftretenden Lagerungsschwindel z. B. steht es als Lagerungstraining ganz im Vordergrund, aber auch bei anderen Ursachen des Schwindels kann Training die Beschwerden wesentlich bessern. Nach dem Motto "Was nicht passt, wird passend gemacht" lernt das Gehirn, die (widersprüchlichen) Informationen neu zu verrechnen und korrigiert den Schwindel so selbst. Die verständliche und als Erstmaßnahme aus Sicherheitsgründen durchaus richtige Haltung, sich hinzulegen und zu schonen, vermeidet den Schwindel zwar zunächst einmal, wirkt sich aber auf Dauer ungünstig aus. Einzelheiten, welche Übungen am besten geeignet sind, finden Sie beim jeweiligen Krankheitsbild.
Manchmal ist Schwindel psychisch oder psychosomatisch bedingt oder ein organischer Schwindel wird durch psychische Faktoren verfestigt (Angst vor dem nächsten Schwindelanfall). Hier muss (zusätzlich) die psychische Störung behandelt werden, etwa durch eine Verhaltenstherapie.
Pharmakotherapie
Medikamente gegen Schwindel, Antivertiginosa genannt, sollten nur bei heftigem Drehschwindel mit Übelkeit kurzzeitig eingesetzt werden, bis die quälende Übelkeit abgeklungen ist, z. B bei der Neuritis vestibularis.
Als Erstlinien-Therapie hat sich in internationalen Studien Dimenhydrinat durchgesetzt.
Liegt dem Schwindel eine vestibuläre Störung zugrunde, verordnen manche Ärzte Flunarizin (z. B. Flunavert®). Auch das für den Morbus Menière zugelassene Betahistin (z. B. Vasomotal®) wird bei Schwindel off label (also ohne Zulassung für diesen speziellen Zweck) eingesetzt, auch wenn es bisher in klinischen Studien noch keinen Wirkungsnachweis erbringen konnte.
Die Behandlung des Schwindels im Rahmen der Reisekrankheit wird im entsprechenden Artikel beschrieben.
Behandlung von Altersschwindel
Bei älteren Menschen kann der Arzt häufig keine eindeutige Ursache für den Schwindel feststellen. In diesen Fällen geht man alle möglicherweise beteiligten Störfaktoren an, auch wenn jeder für sich allein nicht ausreicht, den Schwindel zu erklären. So werden die eingenommenen Medikamente überprüft, Augen und Ohren bzw. Brille und Hörgerät kontrolliert und Blutdruck und Blutzucker optimal eingestellt. Gleichzeitig wird durch Hilfsmittel, wie etwa Gehhilfen und Haltemöglichkeiten, im häuslichen Umfeld versucht, die Sturzgefahr zu mindern. Physiotherapie und Trainingsprogramme haben einen (mindestens) genauso hohen Stellenwert wie bei jüngeren Menschen, erfordern aber nicht selten erhebliche Motivationsarbeit.
Ihr Apotheker empfiehlt
- Lassen Sie jeden Schwindel, der im Alltag zu Stand- und Gangunsicherheit und damit zu Sturz- und Verletzungsgefahr führt, zügig vom Arzt abklären.
- Wenn Sie unter Schwindel leiden, meiden Sie den Auslöser des Schwindels. Ist der Auslöser unbekannt, führen Sie ein Schwindeltagebuch. Auf diese Weise lassen sich die auslösenden Ursachen erkennen.
- Lassen Sie bei neu aufgetretenem Schwindel auch abklären, ob Sie eine Brille brauchen oder ob Ihre Brille angepasst werden muss.
- Halten Sie sich im Schwindelanfall gut fest oder legen Sie sich hin.
- Im akuten Anfall hilft oft ein nasser Lappen auf Stirn oder Nacken und bewusste tiefe Bauchatmung.
- Fahren Sie weder Auto noch Fahrrad, solange Sie unter Schwindel leiden.
Weiterführende Informationen
Dr. Bodo Schiffmann: Jetzt hole ich mir mein Leben zurück, Hilfe zur Selbsthilfe für Schwindelpatienten. Tredition, 2018. Buch mit vielen interessanten Informationen und Anregungen für Schwindelpatienten. Video zum Befreiungsmanöver nach Epley: https://www.youtube.com/watch?v=ePodC5wHR7c Video zum Bfreiungsmanöver nach Semont: https://www.youtube.com/watch?v=3M7HtMdRy3w
Tics
Tics (Ticks): Plötzliche, unwillkürliche, wiederholte, aber unrhythmische Bewegung oder Lauterzeugungen, die sich allenfalls kurzzeitig unterdrücken lassen. Typische Beispiele für motorische Tics sind krampfhaftes, unwillkürliches Augenblinzeln, Grimassieren oder Körperverdrehungen. Vokale Tics äußern sich im Aussprechen obszöner Wörter, Räuspern oder durch Laut-Wiederholungen.
Tic-Störungen. Störungen, bei denen die Betroffenen wiederholt unter Tics leiden. Kindliche Tic-Störungen sind häufig: 15 % aller Kinder im Grundschulalter haben Tics, die im Verlauf von Wochen und Monaten meist wieder verschwinden (transiente Tic-Störungen). Zu den chronischen Tic-Störungen gehört vor allem das Tourette-Syndrom (Gilles-de-la-Tourette-Syndrom). Hierbei handelt es sich um eine neuropsychiatrische Erkrankung mit motorischen und vokalen Tics, die sich im Kindesalter ausbildet und meist ein Leben lang bestehen bleibt. Das Tourette-Syndrom ist bei Männern häufiger als bei Frauen.
Grundlage der Behandlung bei Tic-Störungen sind Informationen und Aufklärung, sowohl des Betroffenen, der Familie als auch des Umfelds. Daneben gibt es verhaltenstherapeutische Ansätze, die bei Tic-Störungen zu einer Besserung führen. Sind die Tics sehr belastend, kommen Medikamente wie Neuroleptika zum Einsatz. Bei schweren, nicht therapierbaren Fällen kann bei Erwachsenen das Einpflanzen eines Hirnschrittmachers erwogen werden.
Symptome und Leitbeschwerden
- Unwillkürliches, wiederholtes Blinzeln, Naserümpfen, Mundöffnen, Räuspern oder Schulterzucken
- Manchmal komplexe Tics, z. B. Körperverdrehung oder das automatische Nachsprechen von gehörten Wörtern
- Bewegungen nur für kurze Zeit willkürlich unterdrückbar, Verstärkung bei Stress
- Oft unangenehmes Vorgefühl vor dem Tic
- Beim Tourette-Syndrom: Beginn häufig mit Blinzel-Tic, dann Entwicklung verschiedener Tics, selten in Verbindung mit dem Gebrauch obszöner Worte oder Gesten.
Wann zum Arzt
In den nächsten Tagen, wenn
- wiederholt oben genannte auffällige Bewegungen auftreten.
Die Erkrankung
Tics werden nach ihrer Qualität (motorisch/vokal) und ihrer Komplexität (einfach/komplex) eingeteilt. Sie treten einzeln oder in Serien auf. Beispiele für Tics sind:
- Einfache motorische Tics: Blinzeln, Stirnrunzeln, Grimassieren, Beißen in die Wangeninnenseite, Kopf schütteln, Fußbewegungen
- Komplexe motorische Tics: Sprünge, Körperverdrehungen, Zupfen an Kleidung, Echopraxie (ungewollte Wiederholung von Bewegungen anderer Menschen), Kopropraxie (unflätige Gesten)
- Einfache vokale Tics: Räuspern, Schmatzen, Bellen, Pfeifen, Summen, Ausrufen von einzelnen Silben
- Komplexe vokale Tics: Echolalie (automatisches, ungewolltes Nachsprechen von Gehörtem), Koprolalie (Aussprechen obszöner Wörter, z. B. aus der Fäkalsprache).
Einfache motorische Tics wie Naserümpfen oder Blinzeln treten bei etwa 15 % aller Kinder im Grundschulalter auf. Meist gibt sich der Tic nach Wochen bis Monaten wieder (begünstigt durch konsequentes Ignorieren), um später noch ein- oder mehrfach zu erscheinen und schließlich ganz zu verschwinden. Solche Tics sind keine Erkrankung und sollten deshalb nicht problematisiert werden. Ärzte nennen dieses Auftreten von Tics auch transiente Tic-Störung.
Tourette-Syndrom und weitere chronische Tic-Störungen
Anders beim Tourette-Syndrom: Anstatt zu verschwinden, kommen neue Tics hinzu, auch vokale Tics mit unwillkürlichen Laut- und Wortäußerungen. Diese vielen Tics, manchmal mit obszöner Bedeutung, werden von der Umgebung oft als Provokation fehlgedeutet und führen dadurch zu Problemen. Das Auftreten von obszönen Gesten und Lauten ist jedoch selten und tritt nur bei 5–10 % der Erkrankten auf. Einige Tics gehen bis zur Selbstverletzung (z. B. durch wiederholtes Beißen in die Wangeninnenseite), eine Gefährdung anderer hingegen ist extrem selten. 80 bis 90 % der Patienten mit einem Tourette-Syndrom leiden zusätzlich an einer Aufmerksamkeitsdefizit-Hyperaktivitätsstörung, bis zu 70 % an Zwängen und bis zu 40 % an Ängsten.
Neben dem Tourette-Syndrom gibt es noch zwei weitere chronische Tic-Störungen: die chronische vokale Tic-Störung und die chronische motorische Tic-Störung. Als Diagnosekriterien gilt bei beiden Störungen, dass sie mindestens ein Jahr andauern und die ersten Symptome vor dem 18. Lebensjahr aufgetreten sind. Die beiden Krankheiten unterscheiden sich darin, dass bei der chronischen vokalen Tic-Störung die Betroffenen nur unter vokalen Tics leiden, bei der chronischen motorischen Tic-Störung dagegen unter motorischen Tics.
Ursachen
Woher Tic-Störungen und das Tourette-Syndrom kommen, ist unklar. Als sicher gilt eine erbliche Veranlagung. Es mehren sich die Hinweise auf eine Botenstoffwechselstörung mit einem Dopaminüberschuss im Gehirn als organische Grundlage, auch Streptokokkeninfektionen werden als mitursächlich diskutiert. Die Störungen betreffen vor allem die Basalganglien im Gehirn, dies sind Regionen, in denen reguliert wird, welchen Impulsen ein Mensch nachgibt und welchen nicht.
Manchmal treten Tics sekundär im Rahmen anderer Erkrankungen auf, z. B. beim Morbus Wilson oder der Huntington Erkrankung. In seltenen Fällen können auch Drogen (Kokain) oder Medikamente Tics auslösen, z. B. Carbamazepin oder Phenytoin.
Diagnosesicherung
Zur Diagnose von Tics und Tic-Störungen genügt dem erfahrenen Arzt meist das klinische Bild, technische Untersuchungen dienen zum Ausschluss anderer Erkrankungen.
Für das Vorliegen eines Tourette-Syndroms müssen folgende Kriterien erfüllt sein:
- Mindestens zwei motorische Tics
- Mindestens ein vokaler Tic
- Krankheitsbeginn in der Kindheit
- Krankheitsdauer mindestens ein Jahr (Unterbrechungen möglich).
Differenzialdiagnosen. Krampfhaftes Blinzeln findet sich z. B. beim Blepharospasmus, ansonsten kommen Muskelzuckungen des Gesichts auch als Spasmus hemifacialis, z. B. durch Druck auf den Gesichtsnerven (N. fazialis) vor. Des Weiteren können Tics auch verwechselt werden mit Zwangshandlungen oder allgemeiner Hyperaktivität.
Behandlung
Eine ursächliche Behandlung der Tic-Störungen und des Tourette-Syndroms ist nicht möglich. Oft ist den Betroffenen schon geholfen, wenn ihre Umgebung über die Erkrankung aufgeklärt wird und ihnen daher mehr Verständnis entgegenbringt.
Stören die Tics sehr, können sie durch Medikamente gelindert werden. In Deutschland verordnen die Ärzte dazu vor allem Neuroleptika wie Tiaprid (z. B. Tiapridex®), Sulpirid (z. B. Dogmatil®), Aripiprazol (Abilify) und Risperidon (Risperdal®). Neuroleptika werden vorsichtig aufdosiert, weil sie eine ganze Reihe von unerwünschten Wirkungen haben.
Verschiedene Formen der Verhaltenstherapie (Reaktionsumkehr-Behandlung, Exposure and responsive prevention) gelten als Behandlungsalternative zu den Medikamenten.
Für Erwachsene mit hohem Leidensdruck und schwerem, mit Medikamenten nicht behandelbarem Tourette-Syndrom, kommt auch die tiefe Hirnstimulation in Frage. Das Einpflanzen eines solchen "Hirnschrittmachers" ist bei Tics allerdings noch nicht so etabliert wie bei der Parkinson-Krankheit und die Ergebnisse sind zum Teil noch widersprüchlich.
Prognose
Transiente Tic-Störungen kommen nur bei Kindern vor, bestehen nur für einige Wochen oder Monate und benötigen meist keine Therapie.
Bei Patienten mit chronischen Tic-Störungen wie dem Tourette-Syndrom verstärken sich die Symptome oft in der Pubertät, danach kommt es jedoch häufig zu einer spontanen Verbesserung.
Ihr Apotheker empfiehlt
- Selbsthilfegruppen. Neben umfassenden Informationen über die Erkrankung ist für die Betroffenen und ihre Familienangehörigen oft der Kontakt zu einer Selbsthilfegruppe hilfreich.
- Schwerbehindertenausweis. Lassen Sie prüfen, ob bei Ihrem Kind mit Tourette-Syndrom die Voraussetzungen für einen Schwerbehindertenausweis gegeben sind. Ab einem gewissen Grad der Behinderung haben die Kinder Anspruch auf Nachteilsausgleiche, z. B. auf spezielle Hilfsmittel für die Schule, Zugabe von Arbeitszeit bei Leistungstests oder auf Begleitung durch eine dritte Person. Ausführliche Informationen dazu gibt es beim knw Kindernetzwerk e. V.
- Pflegeleistungen. Ist der Pflegebedarf Ihres Kindes durch eine Tic-Störung erhöht, haben Sie eventuell Anspruch auf Leistungen aus der Pflegeversicherung.
Weiterführende Informationen
www.tourette-gesellschaft.de – Internetseite der Tourette-Gesellschaft Deutschland e. V. (TGD, Göttingen): Bietet eine große Auswahl an Informationen zum Tourette-Syndrom wie Erfahrungsberichte und Behandlungsmethoden.
Der Interessenverband Tic & Tourette Syndrome bietet Unterstützung und praktische Hilfe für Betroffene. Link zur Webseite: https://iv-ts.de/
Ausführliche Informationen zum Nachteilsausgleich für Kinder mit einer schweren Behinderung finden sich unter https://www.kindernetzwerk.de/downloads/aktiv/2019/2018_Nachteilsausgleich.pdf
Die interessante Biografie des Neurologen Gilles de la Tourette, Erstbeschreiber der Tics, findet sich unter https://www.iv-ts.de/wp-content/uploads/2014/09/biografie.pdf







